library(tidyverse)
library(here)
derivedpath <- here("courses", "foundations-r", "data", "derived")
resultspath <- here("courses", "foundations-r", "results", "modeling", "naive_pooled")
dir.create(resultspath, recursive = TRUE, showWarnings = FALSE)
final_path <- file.path(derivedpath, "analysis_final_locked.csv")
if (!file.exists(final_path)) {
stop("Final locked dataset not found. Render the final dataset assembly lesson first:\n", final_path)
}
analysis <- read_csv(final_path)
obs <- analysis %>% filter(EVID == 0, !is.na(DV), DOSE > 0, is.na(BLQ))Naive Pooled Modeling with nls()
Fit a one-compartment oral PK model using naive pooled data, interpret structural parameters, and understand the limitations of ignoring inter-individual variability.
Tip
Big picture: Before hierarchical modeling, a common first step is a naive pooled fit.
Here, we ignore inter-individual variability and fit one structural model to all subjects simultaneously.
Learning Objectives
By the end of this lesson, you will be able to:
- Define a one-compartment oral PK model using CL/V parameterization.
- Perform a naive pooled nonlinear fit using
nls(). - Interpret ka, CL, V, ke, and half-life from a pooled model.
- Generate pooled fitted curves and diagnostics.
- Explain the limitations of naive pooled modeling.
Key Ideas
- Naive pooling treats all subjects as if they share identical parameters.
- Dose differences still affect predictions through the structural equation.
- Placebo (DOSE = 0) is excluded from fitting because the model describes post-dose PK after an administered dose.
- Between-subject variability is absorbed into residual error.
- Parameter estimates represent an “average structural subject.”
- This approach is simple but statistically limited.
Setup
Worked Example 1: Structural Model (CL/V Parameterization)
First, we define the concentration-time function that nls() will use during estimation.
\[ C(t) = \frac{D \cdot k_a}{V (k_a - k)} \left(e^{-k t} - e^{-k_a t}\right), \quad k = \frac{CL}{V}. \]
one_comp_cl <- function(time, dose, ka, CL, V) {
k <- CL / V
(dose * ka / (V * (ka - k))) *
(exp(-k * time) - exp(-ka * time))
}Worked Example 2: Fit Naive Pooled Model
Now we fit one shared set of structural parameters to all non-placebo observations.
naive_fit <- nls(
DV ~ one_comp_cl(TIME, DOSE, ka, CL, V),
data = obs,
start = list(ka = 1, CL = 5, V = 50),
control = nls.control(maxiter = 200, warnOnly = TRUE)
)
summary(naive_fit)
Formula: DV ~ one_comp_cl(TIME, DOSE, ka, CL, V)
Parameters:
Estimate Std. Error t value Pr(>|t|)
ka 1.3787 0.1649 8.361 2.75e-15 ***
CL 6.2273 0.4359 14.287 < 2e-16 ***
V 68.3905 3.4250 19.968 < 2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.1905 on 286 degrees of freedom
Number of iterations to convergence: 5
Achieved convergence tolerance: 8.536e-07Interpretation:
- One ka, CL, V shared across all subjects.
- Differences in profiles arise only from DOSE and residual error.
Worked Example 3: Convert to Interpretable Parameters
The fitted coefficients are useful, but derived quantities such as elimination rate and half-life are easier to interpret.
est <- coef(naive_fit)
ka_pop <- est["ka"]
CL_pop <- est["CL"]
V_pop <- est["V"]
ke_pop <- CL_pop / V_pop
half_life <- log(2) / ke_pop
tibble(
ka = ka_pop,
CL = CL_pop,
V = V_pop,
ke = ke_pop,
half_life = half_life
)# A tibble: 1 × 5
ka CL V ke half_life
<dbl> <dbl> <dbl> <dbl> <dbl>
1 1.38 6.23 68.4 0.0911 7.61Worked Example 4: Visual Diagnostics
Predictions by subject show where the pooled model follows the general trend and where it misses individual behavior.
Generate predictions:
obs <- obs %>%
mutate(pred = predict(naive_fit))Plot by subject:
p <- ggplot(obs, aes(TIME, DV)) +
geom_point(alpha = 0.6) +
geom_line(aes(y = pred, group = ID), color = "blue") +
facet_wrap(~ ID) +
labs(
title = "Naive Pooled Fit by Subject",
x = "Time After Dose (h)",
y = "Concentration"
)
p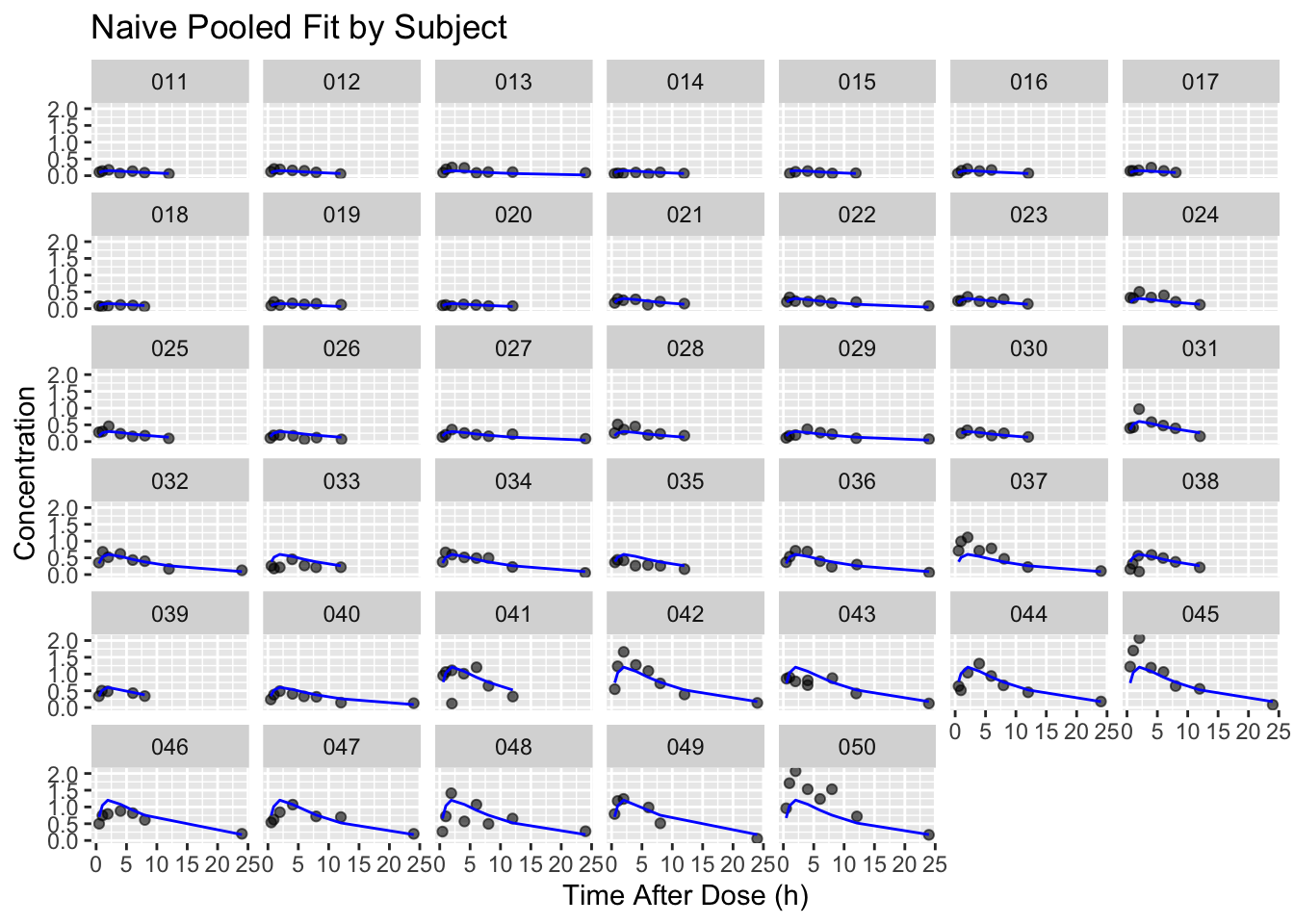
Save:
ggsave(file.path(resultspath, "naive_pooled_fits.png"), p, width = 10, height = 8)Worked Example 6: Residual Diagnostics
Residual plots help separate random scatter from systematic patterns that suggest model limitations.
obs <- obs %>%
mutate(residual = DV - pred)
ggplot(obs, aes(pred, residual)) +
geom_point(alpha = 0.6) +
geom_hline(yintercept = 0, linetype = 2) +
labs(
title = "Residuals vs Fitted (Naive Pooled)",
x = "Fitted",
y = "Residual"
)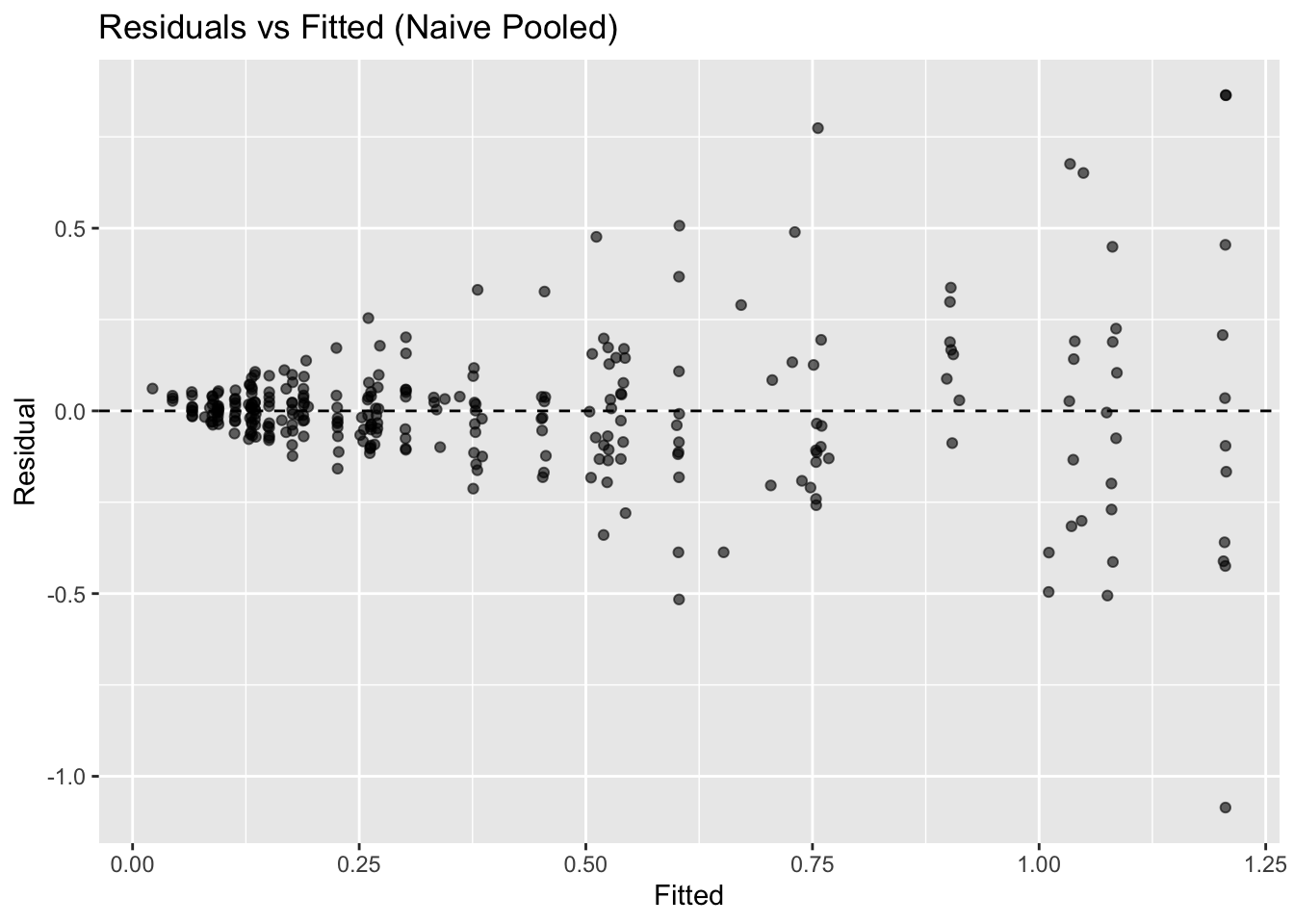
What to look for:
- Systematic curvature → structural misspecification.
- Increasing spread → heteroscedasticity.
- Clustering by subject → ignored inter-individual variability.
In this fit, the residual spread increases at higher fitted concentrations, suggesting heteroscedasticity where variability grows with concentration magnitude. In practice, this often motivates log-transformed modeling or proportional error structures, which are handled more naturally in mixed-effects frameworks such as nlme and nlmixr2.
Warning
- Naive pooled models underestimate true variability.
- Residual error absorbs between-subject differences.
- Do not interpret this as a population PK model.
Strategies
- Work from the locked dataset (
analysis_final_locked.csv). - Filter to observation rows (
EVID == 0). - Use reasonable starting values.
- Inspect residual patterns carefully.
- Treat results as exploratory, not definitive.
Common Mistakes
- Including placebo rows in a model that assumes a nonzero administered dose.
- Interpreting naive pooled estimates as population mixed-effects estimates.
- Trusting convergence without checking fitted curves and residuals.
- Treating between-subject differences as residual noise only.
- Using the pooled model for decisions without acknowledging its limitations.
Practice Problems
- Stratify residuals by
DOSE— do higher doses show larger error? - Create a log-scale concentration plot.
- Compute RMSE of the pooled fit.
TipStep-by-Step Solutions
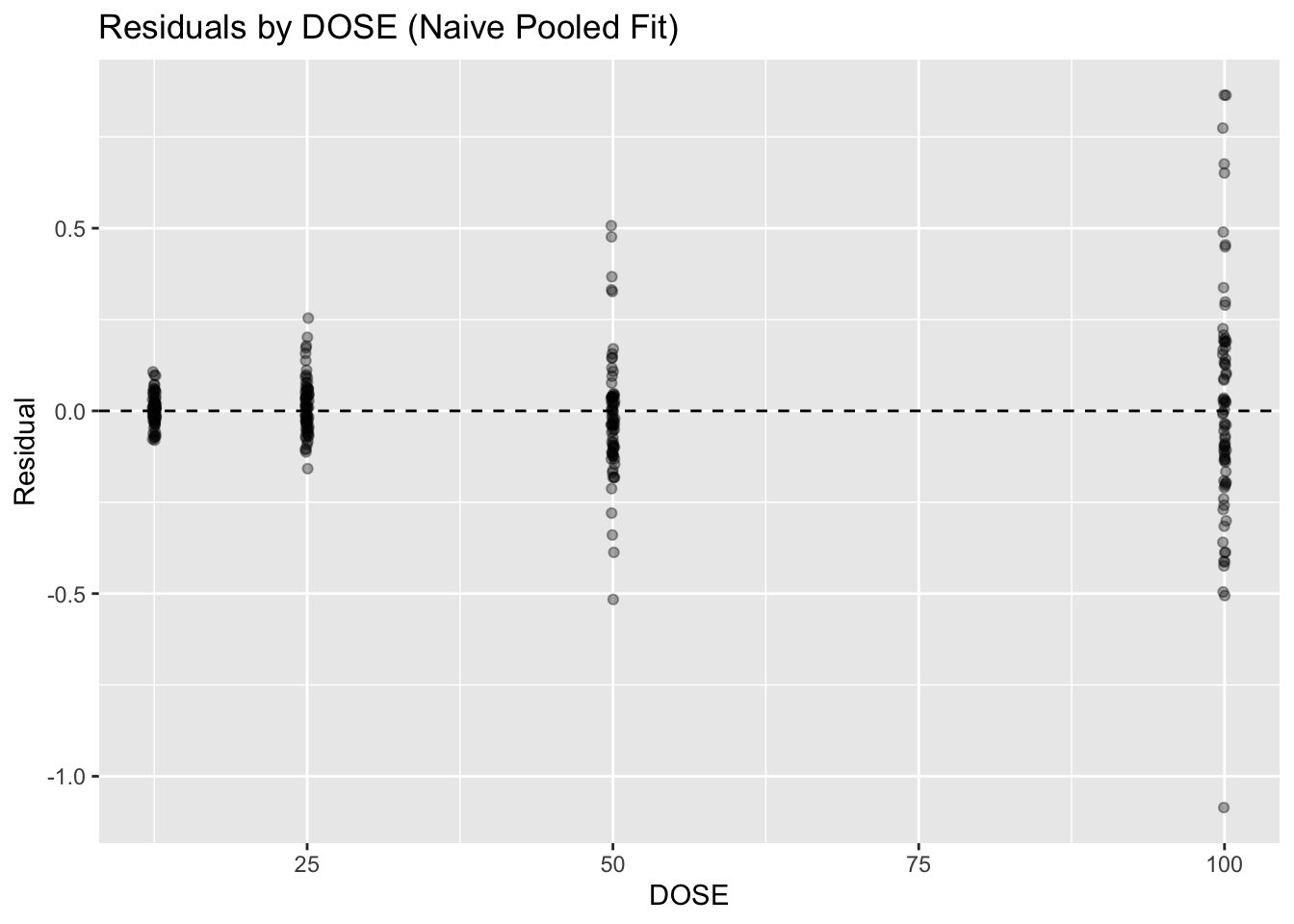
Problem 1: Stratify residuals by DOSE
This checks whether residual magnitude changes with dose. Larger residuals at higher doses can suggest heteroscedasticity, scaling issues, or structural model misspecification.
obs %>%
group_by(DOSE) %>%
summarise(
n = n(),
rmse = sqrt(mean(residual^2, na.rm = TRUE)),
median_abs_resid = median(abs(residual), na.rm = TRUE),
.groups = "drop"
) %>%
arrange(DOSE)# A tibble: 4 × 4
DOSE n rmse median_abs_resid
<dbl> <int> <dbl> <dbl>
1 12.5 67 0.0428 0.0242
2 25 72 0.0774 0.0478
3 50 74 0.166 0.0854
4 100 76 0.320 0.170 A quick visual:
ggplot(obs, aes(DOSE, residual)) +
geom_point(alpha = 0.35, position = position_jitter(width = 0.15)) +
geom_hline(yintercept = 0, linetype = 2) +
labs(
title = "Residuals by DOSE (Naive Pooled Fit)",
x = "DOSE",
y = "Residual"
)
Problem 2: Create a log-scale concentration plot
For a log-scale plot, we filter to positive concentrations because scale_y_log10() cannot display zero or negative values.
p_log <- obs %>%
filter(!is.na(DV) & DV > 0) %>%
ggplot(aes(TIME, DV, group = ID)) +
geom_line(alpha = 0.25) +
geom_point(size = 0.7) +
facet_wrap(~ DOSE) +
scale_y_log10() +
labs(
title = "Observed Concentration–Time Profiles by DOSE (Log Scale)",
x = "Time After Dose (h)",
y = "Concentration (log scale)"
)
p_log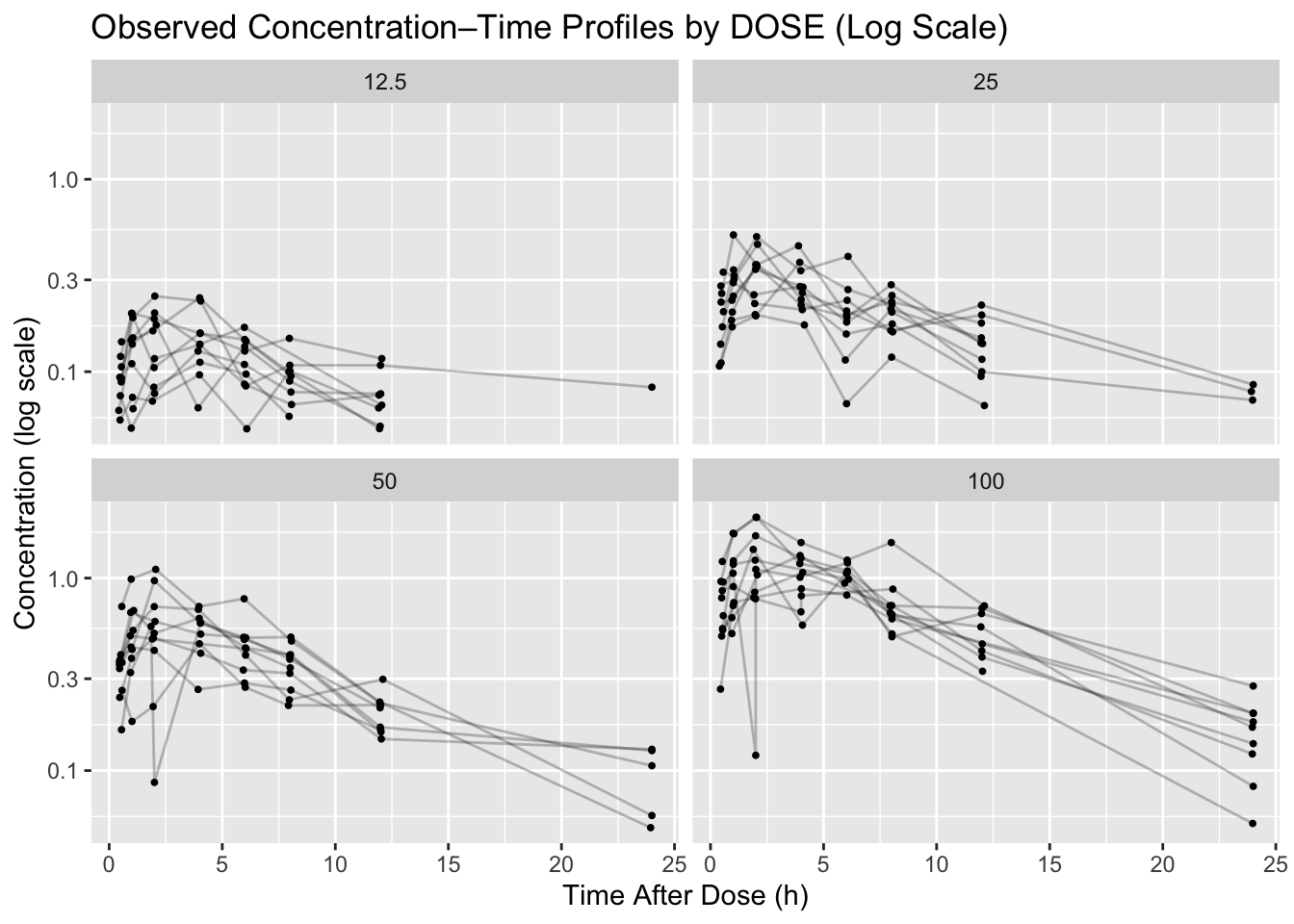
Save it:
ggsave(file.path(resultspath, "profiles_by_dose_log_observed.png"), p_log, width = 9, height = 6)Problem 3: Compute RMSE of the pooled fit
rmse <- sqrt(mean(obs$residual^2, na.rm = TRUE))
rmse[1] 0.1895263You can also compute RMSE by dose, which is often more informative than one pooled value:
obs %>%
group_by(DOSE) %>%
summarise(
rmse = sqrt(mean(residual^2, na.rm = TRUE)),
.groups = "drop"
) %>%
arrange(DOSE)# A tibble: 4 × 2
DOSE rmse
<dbl> <dbl>
1 12.5 0.0428
2 25 0.0774
3 50 0.166
4 100 0.320 Summary
- You fit a one-compartment oral model using naive pooling.
- One parameter set describes all subjects.
- Dose explains systematic profile differences.
- Residual error absorbs inter-individual variability.
- This approach is simple but limited.
TipQuick Tips
- Naive pooling is a useful baseline, not a final answer.
- Always facet plots by subject to reveal hidden variability.
- Large residual spread often signals ignored hierarchy.
- CL/V parameterization keeps interpretation physiologically meaningful.
- Move to mixed-effects modeling when variability matters.